გოშეს დაავადება Gaucher’s disease
ეტიოლოგია
გოშეს დაავადება გადაეცემა რეცესიულად; დაავადებული მშობლების ბავშვები, როგორც წესი არ ავადდებიან; თუმცა აღინიშნება ნათესავებს შორის დაავადების შემთხვევები. გენის მუტაცია, რომელიც იწვევს გოშეს დაავადებას, როგორც ჩანს, ხელს უწყობს ამ დეფექტის მქონე პირთა ევოლუციურ გადარჩევას, რამაც განაპირობა ამ მუტაციის გავრცელება ერთი ეთნიკურ ჯგუფს შორის.
პათოგენეზი.
ხდება ლიპიდების – გლუკოცერებროზიდების დაგროვება მაკროფაგებში; მათი გამრავლების ხარჯზე დიდდება ელენთა, ღვიძლი, ირღვევა ლულოვანი ძვლების სტრუქტურა.
კლინიკური სურათი
თავდაპირველად აღინიშნება ელენთის, შემდეგ კი ღვიძლის უსიმპტომო გადიდება, ტკივილი ძვლებში; სისხლში თანდათანობით მატულობს ციტოპენია; ძვლის ტვინში, ღვიძლსა და ელენთაში აღინიშნება გოშეს უჯრედების დიდი რაოდენობა.
დიაგნოზის დადგენა ხდება გოშეს სპეციფიკური უჯრედების აღმოჩენით ელენთის პუნქტატში ან ძვლის ტვინში.
მკურნალობა ავთვისებიანი ფორმის დროს სიმპტომატურია; კეთილთვისებიანი ფორმის დროს გამოხატული თრომბოციტოპენიის შემთხვევაში, კანქვეშა სისხლჩაქცევების ან ელენთის მნიშვნელოვანი გადიდებისას, ნაჩვენებია სპლენექტომია, ძვლის ტვინის ტრანსპლანტაცია.
პროგნოზი
პროგნოზი ავთვისებიანი ფორმის დროს არაკეთილსაიმედოა – ბავშვები იღუპებიან 1 – 2 წლის განმავლობაში, კეთილთვისებიანი ფორმის დროს ავადმყოფთა უმრავლესობა ცოცხლობს ღრმა მოხუცებულობამდე. პროფილაქტიკა: ოჯახში, სადაც უკვე ავადაა ერთი ბავშვი, შესაძლებელია გლუკოცერებროზიდაზას დეფიციტის დიაგნოსტიკა ამნიონის სითხის უჯრედებში, ამასთან რეკომენდებულია ორსულობისშეწყვეტა.
სოფია კიკნაძის FB პრეზენტაცია: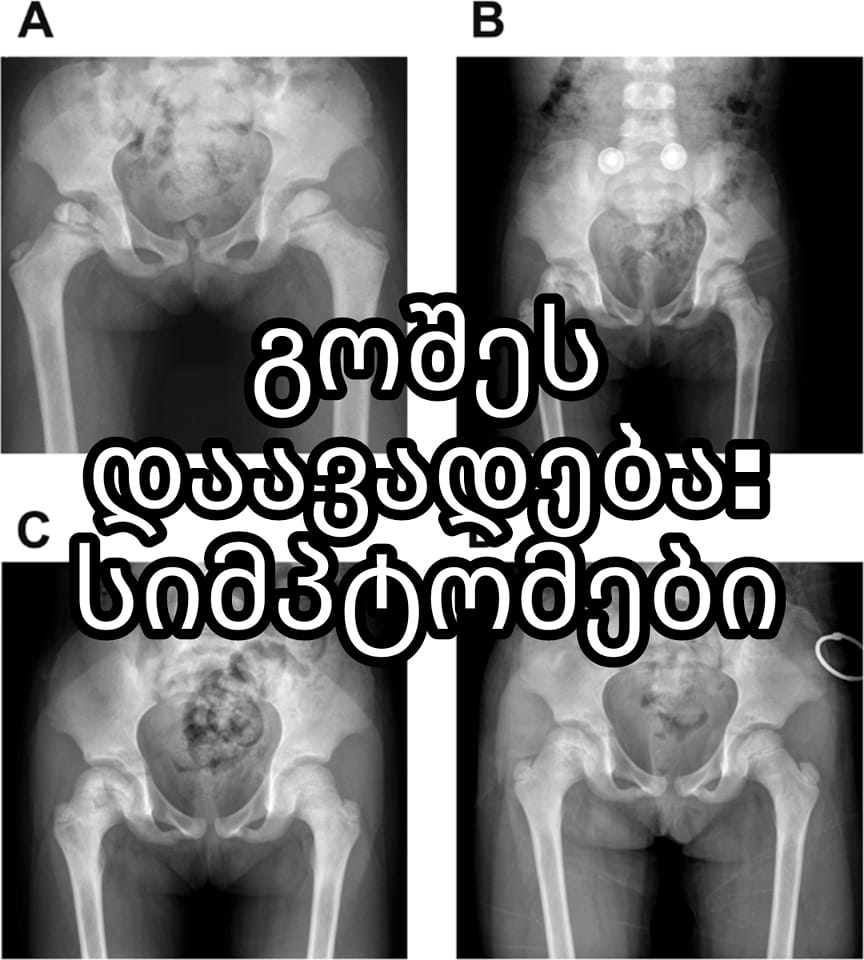
- ჰეპატოსპლენომეგალიას
- ტვინის ფართოდ პროგრესირებად დაზიანებას
- თვალის დისმოტიურობას, სპასტიურობას
- კრუნჩხვებს და კიდურების სიმტკიცეს.
ავადმყოფი ბავშვები ცუდად ყლაპავენ, ჩვეულებრივ იღუპებიან ერთიდან ორ წლამდე. შემთხვევების სიხშირეა 1/100000, არ არსებობს ეთნიკური მიდრეკილება.
გაიგეთ მეტი: https://www.medgeo.net/2009/07/01/gauchers_disease/
გამოგვყევით სოც. ქსელებში:
ლიტერატურა, წყაროები, გაფრთხილება
- გაფრთხილება
- დათეშიძე ლალი, შენგელია არჩილ, შენგელია ვასილ. “ქართული სამედიცინო ენციკლოპედია”. თბილისი, 2005. “ტექინფორმის” დეპონენტი N: 1247. თეიმურაზ ჩიგოგიძის რედაქციით.
- დათეშიძე ლალი, შენგელია არჩილ, შენგელია ვასილ; “ქართული სამედიცინო ენციკლოპედია”. მეორე დეპო-გამოცემა. ჟურნალი “ექსპერიმენტული და კლინიკური მედიცინა”. N: 28. 2006. დეპონენტი პროფესორ თეიმურაზ ჩიგოგიძის საერთო რედაქციით.
2. ლალი დათეშიძე, არჩილ შენგელია. სამედიცინო ენციკლოპედიური განმარტებითი ლექსიკონი
გოშეს დაავადება – ლიპიდოზი: ხასიათდება მეტაბოლიზმის მემკვიდრეობითი დეფექტით გლიკოლიპიდის – ცერებროზიდის ჭარბი დაგროვებითა და მისი შემცველი სპეციფიკური გოშეს უჯრედების ზრდით ლიმფორეტიკულურ სისტემასა და ძვლის ტვინში. ბავშვებში დაავადება ლეტალობით მთავრდება თავის ტვინში ცერებროზიდების დაგროვების გამო. მოზრდილებს აღენიშნებათ სპლენომეგალია, ჰემორაგიული დიათეზი და ცვლილებები ძვლებში. აღინიშნება ტკივილები ძვლებში, პათოლოგიური მოტეხილობები, ბარძაყის ძვლის თავის ასეპტიკური ნეკროზი, ხერხემლის მალების კომპრესია, აგრეთვე ოსტეომიელიტი ან ფსევდოოსტეომიელიტი. დიაგნოზი დგინდება დამახასიათებელი კლინიკური ნიშნებითა და რენტგენოლოგიური კვლევით. სპეციფიკური მკურნალობა – ფერმენტ გლიკოცერებროზიდაზის დანიშვნა.